Brief introduction of metid
Xiaotao Shen PhD (https://www.shenxt.info/)
Created on 2020-03-28 and updated on 2022-09-19
Source:vignettes/metid_introduction.Rmd
metid_introduction.RmdIntroduction & Installation
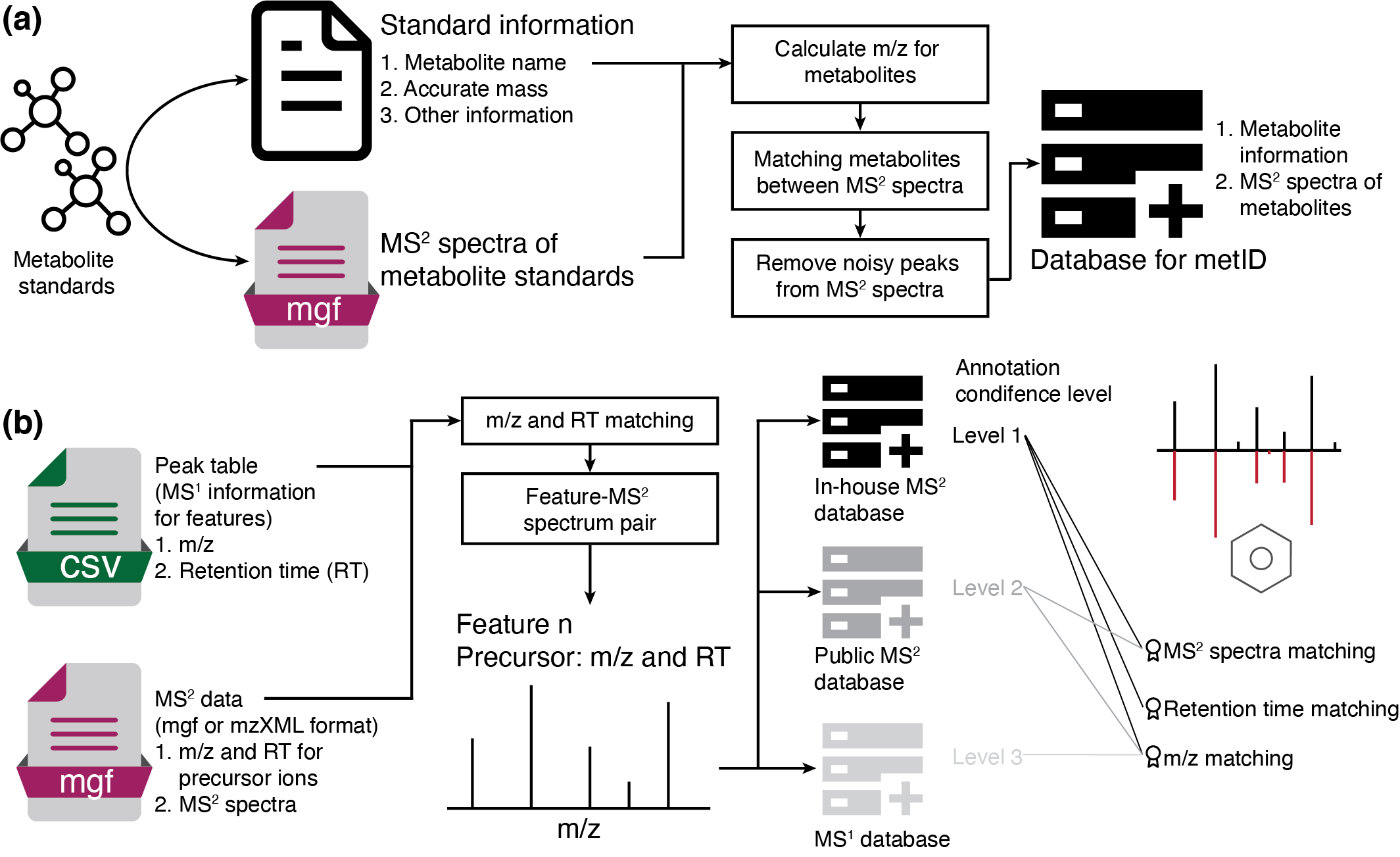
metid is a R package which can be used for in-house (MS2) database construction, and metabolite identification based on in-house and public MS1/MS2 database.
Please install it via github.
if(!require(devtools)){
install.packages("devtools")
}
devtools::install_github("tidymass/metid")All the demo data for metid are from
demoData, so please install it first.
devtools::install_github("jaspershen/demoData")metid is a part of tidymass, so you can
also install it by installing tidymass.
Database construction
Please refer to this article: Construct in-house MS2 datbase using metid for database construction.
Public databases
We have provide some public databases and our in-house databases, so
if you want to use them, please refer this article, Database
provided for metid.
Metabolite identification
If you want to identify metabolite without MS2 spectra, please refer to this article, Annotate metabolites according to MS1 database using metid.
If you want to identify metabolite with MS2 spectra, please refer to this article, Annotate metabolites according to MS2 database using metid.
If you want to identify a peak table with multiple databases, please refer to this article, Identify peak tables with multiple databases.
If you just want to identify single peak, please refer to this article: Identify single peak with metid.
Other tools
metid package also has some useful tools, please refer
to this article, Other
tools in metid