Annotate metabolites according to MS1 database using metid package
Xiaotao Shen PhD (https://www.shenxt.info/)
Created on 2020-03-28 and updated on 2022-09-19
Source:vignettes/metabolite_annotation_using_MS1.Rmd
metabolite_annotation_using_MS1.RmdMS1 data preparation
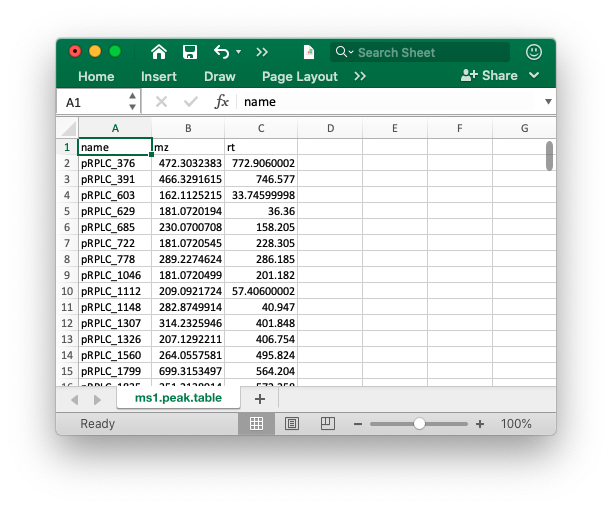
The peak table must contain “name” (peak name), “mz” (mass to charge ratio) and “rt” (retention time, unit is second). It can be from any data processing software (XCMS, MS-DIAL and so on).
Database
The database must be generated using constructDatabase()
function. You can also use the public databases we provoded here.
Data organization
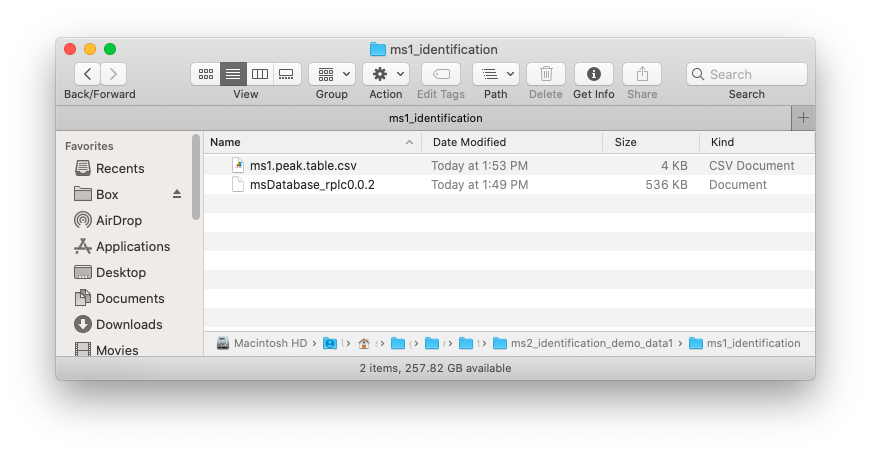
Place the MS1 peak table and databases which you want to use in one folder like below figure shows:
Run identify_metabolites()
function
We use the demo data in metid package to show how to use
metid to identify metabolites without MS2 spectra.
Load demo data
First we load the MS1 peak and database from metid
package and then put them in a example folder.
##create a folder named as example
path <- file.path(".", "example")
dir.create(path = path, showWarnings = FALSE)
##get MS1 peak table from metid
ms1_peak <- system.file("ms1_peak", package = "metid")
file.copy(from = file.path(ms1_peak, "ms1.peak.table.csv"),
to = path, overwrite = TRUE, recursive = TRUE)
#> [1] TRUE
##get database from metid
data("snyder_database_rplc0.0.3", package = "metid")
save(snyder_database_rplc0.0.3, file = file.path(path, "snyder_database_rplc0.0.3"))Now in your ./example, there are two files, namely
ms1.peak.table.csv and msDatabase_rplc_0.0.2,
respectively.
Only use m/z for metabolite identification
First, we only use m/z for metabolite identification.
annotate_result1 <-
identify_metabolites(ms1.data = "ms1.peak.table.csv",
ms1.match.ppm = 15,
rt.match.tol = 1000000,
polarity = "positive",
column = "rp",
path = path,
candidate.num = 3,
database = "snyder_database_rplc0.0.3",
threads = 5)
#>
|
| | 0%
|
|= | 1%
|
|= | 2%
|
|== | 3%
|
|=== | 4%
|
|==== | 5%
|
|==== | 6%
|
|===== | 7%
|
|====== | 8%
|
|====== | 9%
|
|======= | 10%
|
|======== | 11%
|
|======== | 12%
|
|========= | 13%
|
|========== | 14%
|
|========== | 15%
|
|=========== | 16%
|
|============ | 17%
|
|============= | 18%
|
|============= | 19%
|
|============== | 20%
|
|=============== | 21%
|
|=============== | 22%
|
|================ | 23%
|
|================= | 24%
|
|================== | 25%
|
|================== | 26%
|
|=================== | 27%
|
|==================== | 28%
|
|==================== | 29%
|
|===================== | 30%
|
|====================== | 31%
|
|====================== | 32%
|
|======================= | 33%
|
|======================== | 34%
|
|======================== | 35%
|
|========================= | 36%
|
|========================== | 37%
|
|=========================== | 38%
|
|=========================== | 39%
|
|============================ | 40%
|
|============================= | 41%
|
|============================= | 42%
|
|============================== | 43%
|
|=============================== | 44%
|
|================================ | 45%
|
|================================ | 46%
|
|================================= | 47%
|
|================================== | 48%
|
|================================== | 49%
|
|=================================== | 50%
|
|==================================== | 51%
|
|==================================== | 52%
|
|===================================== | 53%
|
|====================================== | 54%
|
|====================================== | 55%
|
|======================================= | 56%
|
|======================================== | 57%
|
|========================================= | 58%
|
|========================================= | 59%
|
|========================================== | 60%
|
|=========================================== | 61%
|
|=========================================== | 62%
|
|============================================ | 63%
|
|============================================= | 64%
|
|============================================== | 65%
|
|============================================== | 66%
|
|=============================================== | 67%
|
|================================================ | 68%
|
|================================================ | 69%
|
|================================================= | 70%
|
|================================================== | 71%
|
|================================================== | 72%
|
|=================================================== | 73%
|
|==================================================== | 74%
|
|==================================================== | 75%
|
|===================================================== | 76%
|
|====================================================== | 77%
|
|======================================================= | 78%
|
|======================================================= | 79%
|
|======================================================== | 80%
|
|========================================================= | 81%
|
|========================================================= | 82%
|
|========================================================== | 83%
|
|=========================================================== | 84%
|
|============================================================ | 85%
|
|============================================================ | 86%
|
|============================================================= | 87%
|
|============================================================== | 88%
|
|============================================================== | 89%
|
|=============================================================== | 90%
|
|================================================================ | 91%
|
|================================================================ | 92%
|
|================================================================= | 93%
|
|================================================================== | 94%
|
|================================================================== | 95%
|
|=================================================================== | 96%
|
|==================================================================== | 97%
|
|===================================================================== | 98%
|
|===================================================================== | 99%
|
|======================================================================| 100%Note: because here we only want to use m/z for metabolite identification, so please set
rt.match.tol(second) > 10,000, for example ‘1000000’ here, so the RT will not be used for filtering.
Other parameters:
ms1.data: The ms1 peak table name.ms1.match.ppm: MS1 match tolerance (ppm).polarity: positive or negative.column: hilic or rp.path: Where are your data placaed?candidate.num: The candidate number for each peak.database: The database name or database.threads: How many threads you want to use.
The return result annotate_result1 is a
metIdentifyClass object, you can directory get the brief
information by print it in console:
annotate_result1
#> --------------metid version-----------
#> 1.0.0
#> -----------Identifications------------
#> (Use get_identification_table() to get identification table)
#> There are 100 peaks
#> 0 peaks have MS2 spectra
#> There are 98 metabolites are identified
#> There are 55 peaks with identification
#> -----------Parameters------------
#> (Use get_parameters() to get all the parameters of this processing)
#> Polarity: positive
#> Collision energy: all
#> database: snyder_database_rplc0.0.3
#> Total score cutoff: 0.5
#> Column: rp
#> Adduct table:
#> (M+H)+;(M+H-H2O)+;(M+H-2H2O)+;(M+NH4)+;(M+Na)+;(M-H+2Na)+;(M-2H+3Na)+;(M+K)+;(M-H+2K)+;(M-2H+3K)+;(M+CH3CN+H)+;(M+CH3CN+Na)+;(2M+H)+;(2M+NH4)+;(2M+Na)+;(2M+K)+;(M+HCOO+2H)+Note:
now we can also provide “databaseClass” object for “database” argument. For example: we load the database first.
snyder_database_rplc0.0.3
#> -----------Base information------------
#> Version: 0.0.2
#> Source: MS
#> Link: http://snyderlab.stanford.edu/
#> Creater: Xiaotao Shen ( shenxt1990@163.com )
#> With RT information
#> -----------Spectral information------------
#> There are 14 items of metabolites in database:
#> Lab.ID; Compound.name; mz; RT; CAS.ID; HMDB.ID; KEGG.ID; Formula; mz.pos; mz.neg; Submitter; Family; Sub.pathway; Note
#> There are 833 metabolites in total
#> There are 356 MS2 spectra in positive mode.
#> There are 534 MS2 spectra in negative mode.
#> Collision energy in positive mode (number:):
#> Total number: 2
#> NCE25; NCE50
#> Collision energy in negative mode:
#> Total number: 2
#> NCE25; NCE50
#> Then we can directory provide this database to
identify_metabolites():
annotate_result2 <-
identify_metabolites(ms1.data = "ms1.peak.table.csv",
ms1.match.ppm = 15,
rt.match.tol = 1000000,
polarity = "positive",
column = "rp",
path = path,
candidate.num = 3,
database = snyder_database_rplc0.0.3,
threads = 5)
#>
|
| | 0%
|
|= | 1%
|
|= | 2%
|
|== | 3%
|
|=== | 4%
|
|==== | 5%
|
|==== | 6%
|
|===== | 7%
|
|====== | 8%
|
|====== | 9%
|
|======= | 10%
|
|======== | 11%
|
|======== | 12%
|
|========= | 13%
|
|========== | 14%
|
|========== | 15%
|
|=========== | 16%
|
|============ | 17%
|
|============= | 18%
|
|============= | 19%
|
|============== | 20%
|
|=============== | 21%
|
|=============== | 22%
|
|================ | 23%
|
|================= | 24%
|
|================== | 25%
|
|================== | 26%
|
|=================== | 27%
|
|==================== | 28%
|
|==================== | 29%
|
|===================== | 30%
|
|====================== | 31%
|
|====================== | 32%
|
|======================= | 33%
|
|======================== | 34%
|
|======================== | 35%
|
|========================= | 36%
|
|========================== | 37%
|
|=========================== | 38%
|
|=========================== | 39%
|
|============================ | 40%
|
|============================= | 41%
|
|============================= | 42%
|
|============================== | 43%
|
|=============================== | 44%
|
|================================ | 45%
|
|================================ | 46%
|
|================================= | 47%
|
|================================== | 48%
|
|================================== | 49%
|
|=================================== | 50%
|
|==================================== | 51%
|
|==================================== | 52%
|
|===================================== | 53%
|
|====================================== | 54%
|
|====================================== | 55%
|
|======================================= | 56%
|
|======================================== | 57%
|
|========================================= | 58%
|
|========================================= | 59%
|
|========================================== | 60%
|
|=========================================== | 61%
|
|=========================================== | 62%
|
|============================================ | 63%
|
|============================================= | 64%
|
|============================================== | 65%
|
|============================================== | 66%
|
|=============================================== | 67%
|
|================================================ | 68%
|
|================================================ | 69%
|
|================================================= | 70%
|
|================================================== | 71%
|
|================================================== | 72%
|
|=================================================== | 73%
|
|==================================================== | 74%
|
|==================================================== | 75%
|
|===================================================== | 76%
|
|====================================================== | 77%
|
|======================================================= | 78%
|
|======================================================= | 79%
|
|======================================================== | 80%
|
|========================================================= | 81%
|
|========================================================= | 82%
|
|========================================================== | 83%
|
|=========================================================== | 84%
|
|============================================================ | 85%
|
|============================================================ | 86%
|
|============================================================= | 87%
|
|============================================================== | 88%
|
|============================================================== | 89%
|
|=============================================================== | 90%
|
|================================================================ | 91%
|
|================================================================ | 92%
|
|================================================================= | 93%
|
|================================================================== | 94%
|
|================================================================== | 95%
|
|=================================================================== | 96%
|
|==================================================================== | 97%
|
|===================================================================== | 98%
|
|===================================================================== | 99%
|
|======================================================================| 100%But what should be noticed is that it have different name for database in the final result:
annotate_result1@database
#> [1] "snyder_database_rplc0.0.3"
annotate_result2@database
#> [1] "MS_0.0.2"It is because that if you give the
databaseClass, soidentify_metabolitescan know the name of database, if just use thesourceandversionas the name for database.
paste(snyder_database_rplc0.0.3@database.info$Source,
snyder_database_rplc0.0.3@database.info$Version,
sep = "_")
#> [1] "MS_0.0.2"Only use m/z and RT for metabolite identification
Here we set RT tolerance (rt.match.tol) as 30 s.
annotate_result2 <-
identify_metabolites(ms1.data = "ms1.peak.table.csv",
ms1.match.ppm = 15,
rt.match.tol = 30,
polarity = "positive",
column = "rp",
path = path,
candidate.num = 3,
database = "snyder_database_rplc0.0.3",
threads = 5)
#>
|
| | 0%
|
|= | 1%
|
|= | 2%
|
|== | 3%
|
|=== | 4%
|
|==== | 5%
|
|==== | 6%
|
|===== | 7%
|
|====== | 8%
|
|====== | 9%
|
|======= | 10%
|
|======== | 11%
|
|======== | 12%
|
|========= | 13%
|
|========== | 14%
|
|========== | 15%
|
|=========== | 16%
|
|============ | 17%
|
|============= | 18%
|
|============= | 19%
|
|============== | 20%
|
|=============== | 21%
|
|=============== | 22%
|
|================ | 23%
|
|================= | 24%
|
|================== | 25%
|
|================== | 26%
|
|=================== | 27%
|
|==================== | 28%
|
|==================== | 29%
|
|===================== | 30%
|
|====================== | 31%
|
|====================== | 32%
|
|======================= | 33%
|
|======================== | 34%
|
|======================== | 35%
|
|========================= | 36%
|
|========================== | 37%
|
|=========================== | 38%
|
|=========================== | 39%
|
|============================ | 40%
|
|============================= | 41%
|
|============================= | 42%
|
|============================== | 43%
|
|=============================== | 44%
|
|================================ | 45%
|
|================================ | 46%
|
|================================= | 47%
|
|================================== | 48%
|
|================================== | 49%
|
|=================================== | 50%
|
|==================================== | 51%
|
|==================================== | 52%
|
|===================================== | 53%
|
|====================================== | 54%
|
|====================================== | 55%
|
|======================================= | 56%
|
|======================================== | 57%
|
|========================================= | 58%
|
|========================================= | 59%
|
|========================================== | 60%
|
|=========================================== | 61%
|
|=========================================== | 62%
|
|============================================ | 63%
|
|============================================= | 64%
|
|============================================== | 65%
|
|============================================== | 66%
|
|=============================================== | 67%
|
|================================================ | 68%
|
|================================================ | 69%
|
|================================================= | 70%
|
|================================================== | 71%
|
|================================================== | 72%
|
|=================================================== | 73%
|
|==================================================== | 74%
|
|==================================================== | 75%
|
|===================================================== | 76%
|
|====================================================== | 77%
|
|======================================================= | 78%
|
|======================================================= | 79%
|
|======================================================== | 80%
|
|========================================================= | 81%
|
|========================================================= | 82%
|
|========================================================== | 83%
|
|=========================================================== | 84%
|
|============================================================ | 85%
|
|============================================================ | 86%
|
|============================================================= | 87%
|
|============================================================== | 88%
|
|============================================================== | 89%
|
|=============================================================== | 90%
|
|================================================================ | 91%
|
|================================================================ | 92%
|
|================================================================= | 93%
|
|================================================================== | 94%
|
|================================================================== | 95%
|
|=================================================================== | 96%
|
|==================================================================== | 97%
|
|===================================================================== | 98%
|
|===================================================================== | 99%
|
|======================================================================| 100%Get detailed annotation information
After get the annotation_result, we can get the detailed
information from it.
Get the processing parameters
We can use get_parameters() function to get the detailed
parameters. This is very useful for reproductive analysis for data
analysis.
metid::get_parameters_metid(annotate_result1)
#> # A tibble: 17 × 3
#> Parameter Meaning Value
#> <chr> <chr> <chr>
#> 1 ms1.ms2.match.mz.tol MS1 features & MS spectra matching mz tolerance (… 25
#> 2 ms1.ms2.match.rt.tol MS1 features & MS spectra matching RT tolerance (… 10
#> 3 ms1.match.ppm MS1 match tolerance (ppm) 15
#> 4 ms2.match.ppm MS2 fragment match tolerance (ppm) 30
#> 5 ms2.match.tol MS2 match tolerance 0.5
#> 6 rt.match.tol RT match tolerance (s) 1e+06
#> 7 polarity Polarity posi…
#> 8 ce Collision energy all
#> 9 column Column rp
#> 10 ms1.match.weight MS1 match weight 0.25
#> 11 rt.match.weight RT match weight 0.25
#> 12 ms2.match.weight MS2 match weight 0.5
#> 13 path Work directory ./ex…
#> 14 total.score.tol Total score tolerance 0.5
#> 15 candidate.num Candidate number 3
#> 16 database MS2 database snyd…
#> 17 threads Thread number 5
metid::get_parameters_metid(annotate_result2)
#> # A tibble: 17 × 3
#> Parameter Meaning Value
#> <chr> <chr> <chr>
#> 1 ms1.ms2.match.mz.tol MS1 features & MS spectra matching mz tolerance (… 25
#> 2 ms1.ms2.match.rt.tol MS1 features & MS spectra matching RT tolerance (… 10
#> 3 ms1.match.ppm MS1 match tolerance (ppm) 15
#> 4 ms2.match.ppm MS2 fragment match tolerance (ppm) 30
#> 5 ms2.match.tol MS2 match tolerance 0.5
#> 6 rt.match.tol RT match tolerance (s) 30
#> 7 polarity Polarity posi…
#> 8 ce Collision energy all
#> 9 column Column rp
#> 10 ms1.match.weight MS1 match weight 0.25
#> 11 rt.match.weight RT match weight 0.25
#> 12 ms2.match.weight MS2 match weight 0.5
#> 13 path Work directory ./ex…
#> 14 total.score.tol Total score tolerance 0.5
#> 15 candidate.num Candidate number 3
#> 16 database MS2 database snyd…
#> 17 threads Thread number 5Check what peaks with annotations
Use which_has_identification() function to get what
peaks have annotions.
which_has_identification(annotate_result1) %>%
head()
#> MS1.peak.name MS2.spectra.name
#> 1 pRPLC_376 NA
#> 2 pRPLC_391 NA
#> 3 pRPLC_603 NA
#> 4 pRPLC_629 NA
#> 5 pRPLC_685 NA
#> 6 pRPLC_722 NABecause there are no ms2 data, so the peaks have no MS2 spectra.
Get the identification table
We can use get_identification_table() to get the
identification table.
table1 <-
get_identification_table(annotate_result1,
candidate.num = 3,
type = "old")
table1
#> # A tibble: 100 × 5
#> name mz rt Candidate.number Identification
#> <chr> <dbl> <dbl> <dbl> <chr>
#> 1 pRPLC_376 472. 773. 3 Compound.name:Chenodeoxycholic acid …
#> 2 pRPLC_391 466. 747. 1 Compound.name:C18:0 AC (Stearoylcarn…
#> 3 pRPLC_603 162. 33.7 2 Compound.name:L(-)-Carnitine;CAS.ID:…
#> 4 pRPLC_629 181. 36.4 3 Compound.name:THEOBROMINE;CAS.ID:NA;…
#> 5 pRPLC_685 230. 158. 3 Compound.name:Pyridoxic acid;CAS.ID:…
#> 6 pRPLC_722 181. 228. 3 Compound.name:THEOBROMINE;CAS.ID:NA;…
#> 7 pRPLC_778 289. 286. 0 NA
#> 8 pRPLC_1046 181. 201. 3 Compound.name:THEOBROMINE;CAS.ID:NA;…
#> 9 pRPLC_1112 209. 57.4 3 Compound.name:5-HYDROXYINDOLEACETATE…
#> 10 pRPLC_1148 283. 40.9 0 NA
#> # … with 90 more rowsThe type is set as old. It means the
identifications for each peak is shown as one character and seperated by
{}. And the order is sorted by
Total score.
You can also set type as new to get another
style.
table2 <-
get_identification_table(annotate_result1, candidate.num = 3,
type = "new")
table2
#> # A tibble: 169 × 19
#> name mz rt MS2.spectra.name Candidate.number Compound.name CAS.ID
#> <chr> <dbl> <dbl> <lgl> <lgl> <chr> <chr>
#> 1 pRPLC_376 472. 773. NA NA Chenodeoxycho… 640-7…
#> 2 pRPLC_376 NA NA NA NA CHOLATE NA
#> 3 pRPLC_376 NA NA NA NA Cholic Acid NA
#> 4 pRPLC_391 466. 747. NA NA C18:0 AC (Ste… 1976-…
#> 5 pRPLC_603 162. 33.7 NA NA L(-)-Carnitine NA
#> 6 pRPLC_603 NA NA NA NA L-Carnitine 541-1…
#> 7 pRPLC_629 181. 36.4 NA NA THEOBROMINE NA
#> 8 pRPLC_629 NA NA NA NA 5-Acetylamino… NA
#> 9 pRPLC_629 NA NA NA NA Theophylline NA
#> 10 pRPLC_685 230. 158. NA NA Pyridoxic acid 82-82…
#> # … with 159 more rows, and 12 more variables: HMDB.ID <chr>, KEGG.ID <chr>,
#> # Lab.ID <chr>, Adduct <chr>, mz.error <dbl>, mz.match.score <dbl>,
#> # RT.error <dbl>, RT.match.score <dbl>, CE <chr>, SS <dbl>,
#> # Total.score <dbl>, Database <chr>If you only want to keep one cancidate for each peak. Please set
candiate.num as 1.
table2 <-
get_identification_table(annotate_result1, candidate.num = 2,
type = "new")
table2
#> # A tibble: 141 × 19
#> name mz rt MS2.spectra.name Candidate.number Compound.name CAS.ID
#> <chr> <dbl> <dbl> <lgl> <lgl> <chr> <chr>
#> 1 pRPLC_376 472. 773. NA NA Chenodeoxycho… 640-7…
#> 2 pRPLC_376 NA NA NA NA CHOLATE NA
#> 3 pRPLC_391 466. 747. NA NA C18:0 AC (Ste… 1976-…
#> 4 pRPLC_603 162. 33.7 NA NA L(-)-Carnitine NA
#> 5 pRPLC_603 NA NA NA NA L-Carnitine 541-1…
#> 6 pRPLC_629 181. 36.4 NA NA THEOBROMINE NA
#> 7 pRPLC_629 NA NA NA NA 5-Acetylamino… NA
#> 8 pRPLC_685 230. 158. NA NA Pyridoxic acid 82-82…
#> 9 pRPLC_685 NA NA NA NA Pyridoxic acid 82-82…
#> 10 pRPLC_722 181. 228. NA NA THEOBROMINE NA
#> # … with 131 more rows, and 12 more variables: HMDB.ID <chr>, KEGG.ID <chr>,
#> # Lab.ID <chr>, Adduct <chr>, mz.error <dbl>, mz.match.score <dbl>,
#> # RT.error <dbl>, RT.match.score <dbl>, CE <chr>, SS <dbl>,
#> # Total.score <dbl>, Database <chr>Get the identification information for single peak
We can use get_iden_info() function to get the detailed
information for a sinlge peak. Because it gets the information from the
database, so this function need provide the database.
First, we need to know what peaks have annotations.
which_has_identification(annotate_result1) %>%
head()
#> MS1.peak.name MS2.spectra.name
#> 1 pRPLC_376 NA
#> 2 pRPLC_391 NA
#> 3 pRPLC_603 NA
#> 4 pRPLC_629 NA
#> 5 pRPLC_685 NA
#> 6 pRPLC_722 NAThen we can get the annotation for peak pRPLC_376 use
get_iden_info() function.
get_iden_info(object = annotate_result1,
which.peak = "pRPLC_376",
database = snyder_database_rplc0.0.3)
#> # A tibble: 3 × 22
#> Compound.name CAS.ID HMDB.ID KEGG.ID Lab.ID Adduct mz.error RT.error
#> <chr> <chr> <chr> <chr> <chr> <chr> <dbl> <lgl>
#> 1 Chenodeoxycholic acid … 640-7… HMDB00… C05466 RPLC_… (M+Na… 0.240 NA
#> 2 CHOLATE NA NA NA RPLC_… (M+CH… 0.300 NA
#> 3 Cholic Acid NA HMDB00… NA RPLC_… (M+CH… 0.452 NA
#> # … with 14 more variables: mz.match.score <dbl>, RT.match.score <lgl>,
#> # Total.score <dbl>, CE <lgl>, SS <dbl>, mz <chr>, RT <dbl>, Formula <chr>,
#> # mz.pos <dbl>, mz.neg <dbl>, Submitter <chr>, Family <chr>,
#> # Sub.pathway <chr>, Note <chr>We can get the detailed information for metabolites in database.
Filter identifications
After we get the annotation result use
identify_metabolites() function. We can also use
filter_identification() function to filter annotations
based on m/z, rt and MS2 match tolerance.
annotate_result2_2 <-
filter_identification(object = annotate_result2,
rt.match.tol = 5)
annotate_result2_2
#> --------------metid version-----------
#> 1.0.0
#> -----------Identifications------------
#> (Use get_identification_table() to get identification table)
#> There are 100 peaks
#> 0 peaks have MS2 spectra
#> There are 9 metabolites are identified
#> There are 6 peaks with identification
#> -----------Parameters------------
#> (Use get_parameters() to get all the parameters of this processing)
#> Polarity: positive
#> Collision energy: all
#> database: snyder_database_rplc0.0.3
#> Total score cutoff: 0.5
#> Column: rp
#> Adduct table:
#> (M+H)+;(M+H-H2O)+;(M+H-2H2O)+;(M+NH4)+;(M+Na)+;(M-H+2Na)+;(M-2H+3Na)+;(M+K)+;(M-H+2K)+;(M-2H+3K)+;(M+CH3CN+H)+;(M+CH3CN+Na)+;(2M+H)+;(2M+NH4)+;(2M+Na)+;(2M+K)+;(M+HCOO+2H)+
annotate_result2
#> --------------metid version-----------
#> 1.0.0
#> -----------Identifications------------
#> (Use get_identification_table() to get identification table)
#> There are 100 peaks
#> 0 peaks have MS2 spectra
#> There are 47 metabolites are identified
#> There are 24 peaks with identification
#> -----------Parameters------------
#> (Use get_parameters() to get all the parameters of this processing)
#> Polarity: positive
#> Collision energy: all
#> database: snyder_database_rplc0.0.3
#> Total score cutoff: 0.5
#> Column: rp
#> Adduct table:
#> (M+H)+;(M+H-H2O)+;(M+H-2H2O)+;(M+NH4)+;(M+Na)+;(M-H+2Na)+;(M-2H+3Na)+;(M+K)+;(M-H+2K)+;(M-2H+3K)+;(M+CH3CN+H)+;(M+CH3CN+Na)+;(2M+H)+;(2M+NH4)+;(2M+Na)+;(2M+K)+;(M+HCOO+2H)+Session information
sessionInfo()
#> R version 4.2.1 (2022-06-23)
#> Platform: x86_64-apple-darwin17.0 (64-bit)
#> Running under: macOS Big Sur ... 10.16
#>
#> Matrix products: default
#> BLAS: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRblas.0.dylib
#> LAPACK: /Library/Frameworks/R.framework/Versions/4.2/Resources/lib/libRlapack.dylib
#>
#> locale:
#> [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] tinytools_0.9.1 forcats_0.5.1.9000 stringr_1.4.0 dplyr_1.0.9
#> [5] purrr_0.3.4 readr_2.1.2 tidyr_1.2.0 tibble_3.1.7
#> [9] tidyverse_1.3.1 ggplot2_3.3.6 massdataset_1.0.5 magrittr_2.0.3
#> [13] masstools_0.99.13 metid_1.2.16
#>
#> loaded via a namespace (and not attached):
#> [1] backports_1.4.1 readxl_1.4.0
#> [3] circlize_0.4.15 systemfonts_1.0.4
#> [5] plyr_1.8.7 lazyeval_0.2.2
#> [7] BiocParallel_1.30.3 listenv_0.8.0
#> [9] GenomeInfoDb_1.32.2 Rdisop_1.56.0
#> [11] digest_0.6.29 foreach_1.5.2
#> [13] yulab.utils_0.0.5 htmltools_0.5.2
#> [15] fansi_1.0.3 memoise_2.0.1
#> [17] cluster_2.1.3 doParallel_1.0.17
#> [19] tzdb_0.3.0 openxlsx_4.2.5
#> [21] limma_3.52.2 ComplexHeatmap_2.12.0
#> [23] globals_0.15.1 modelr_0.1.8
#> [25] matrixStats_0.62.0 vroom_1.5.7
#> [27] pkgdown_2.0.5 prettyunits_1.1.1
#> [29] colorspace_2.0-3 rvest_1.0.2
#> [31] haven_2.5.0 textshaping_0.3.6
#> [33] xfun_0.31 crayon_1.5.1
#> [35] RCurl_1.98-1.7 jsonlite_1.8.0
#> [37] impute_1.70.0 iterators_1.0.14
#> [39] glue_1.6.2 gtable_0.3.0
#> [41] zlibbioc_1.42.0 XVector_0.36.0
#> [43] GetoptLong_1.0.5 DelayedArray_0.22.0
#> [45] shape_1.4.6 BiocGenerics_0.42.0
#> [47] scales_1.2.0 vsn_3.64.0
#> [49] DBI_1.1.3 Rcpp_1.0.8.3
#> [51] mzR_2.30.0 viridisLite_0.4.0
#> [53] progress_1.2.2 clue_0.3-61
#> [55] gridGraphics_0.5-1 bit_4.0.4
#> [57] preprocessCore_1.58.0 stats4_4.2.1
#> [59] MsCoreUtils_1.8.0 htmlwidgets_1.5.4
#> [61] httr_1.4.3 RColorBrewer_1.1-3
#> [63] ellipsis_0.3.2 pkgconfig_2.0.3
#> [65] XML_3.99-0.10 dbplyr_2.2.1
#> [67] sass_0.4.1 utf8_1.2.2
#> [69] ggplotify_0.1.0 tidyselect_1.1.2
#> [71] rlang_1.0.3 munsell_0.5.0
#> [73] cellranger_1.1.0 tools_4.2.1
#> [75] cachem_1.0.6 cli_3.3.0
#> [77] generics_0.1.3 broom_1.0.0
#> [79] evaluate_0.15 fastmap_1.1.0
#> [81] mzID_1.34.0 yaml_2.3.5
#> [83] ragg_1.2.2 bit64_4.0.5
#> [85] knitr_1.39 fs_1.5.2
#> [87] zip_2.2.0 ncdf4_1.19
#> [89] pbapply_1.5-0 future_1.26.1
#> [91] xml2_1.3.3 compiler_4.2.1
#> [93] rstudioapi_0.13 plotly_4.10.0
#> [95] png_0.1-7 affyio_1.66.0
#> [97] reprex_2.0.1 bslib_0.3.1
#> [99] stringi_1.7.6 desc_1.4.1
#> [101] MSnbase_2.22.0 lattice_0.20-45
#> [103] ProtGenerics_1.28.0 Matrix_1.4-1
#> [105] ggsci_2.9 vctrs_0.4.1
#> [107] pillar_1.7.0 lifecycle_1.0.1
#> [109] furrr_0.3.0 BiocManager_1.30.18
#> [111] jquerylib_0.1.4 MALDIquant_1.21
#> [113] GlobalOptions_0.1.2 data.table_1.14.2
#> [115] bitops_1.0-7 GenomicRanges_1.48.0
#> [117] R6_2.5.1 pcaMethods_1.88.0
#> [119] affy_1.74.0 IRanges_2.30.0
#> [121] parallelly_1.32.0 codetools_0.2-18
#> [123] MASS_7.3-57 assertthat_0.2.1
#> [125] SummarizedExperiment_1.26.1 rprojroot_2.0.3
#> [127] rjson_0.2.21 withr_2.5.0
#> [129] S4Vectors_0.34.0 GenomeInfoDbData_1.2.8
#> [131] parallel_4.2.1 hms_1.1.1
#> [133] grid_4.2.1 rmarkdown_2.14
#> [135] MatrixGenerics_1.8.1 lubridate_1.8.0
#> [137] Biobase_2.56.0