
About
metid is a R package which is used for metabolite identification based on in-house database and public database based on accurate mass (m/z), retention time (RT) and/or MS2 spectra.
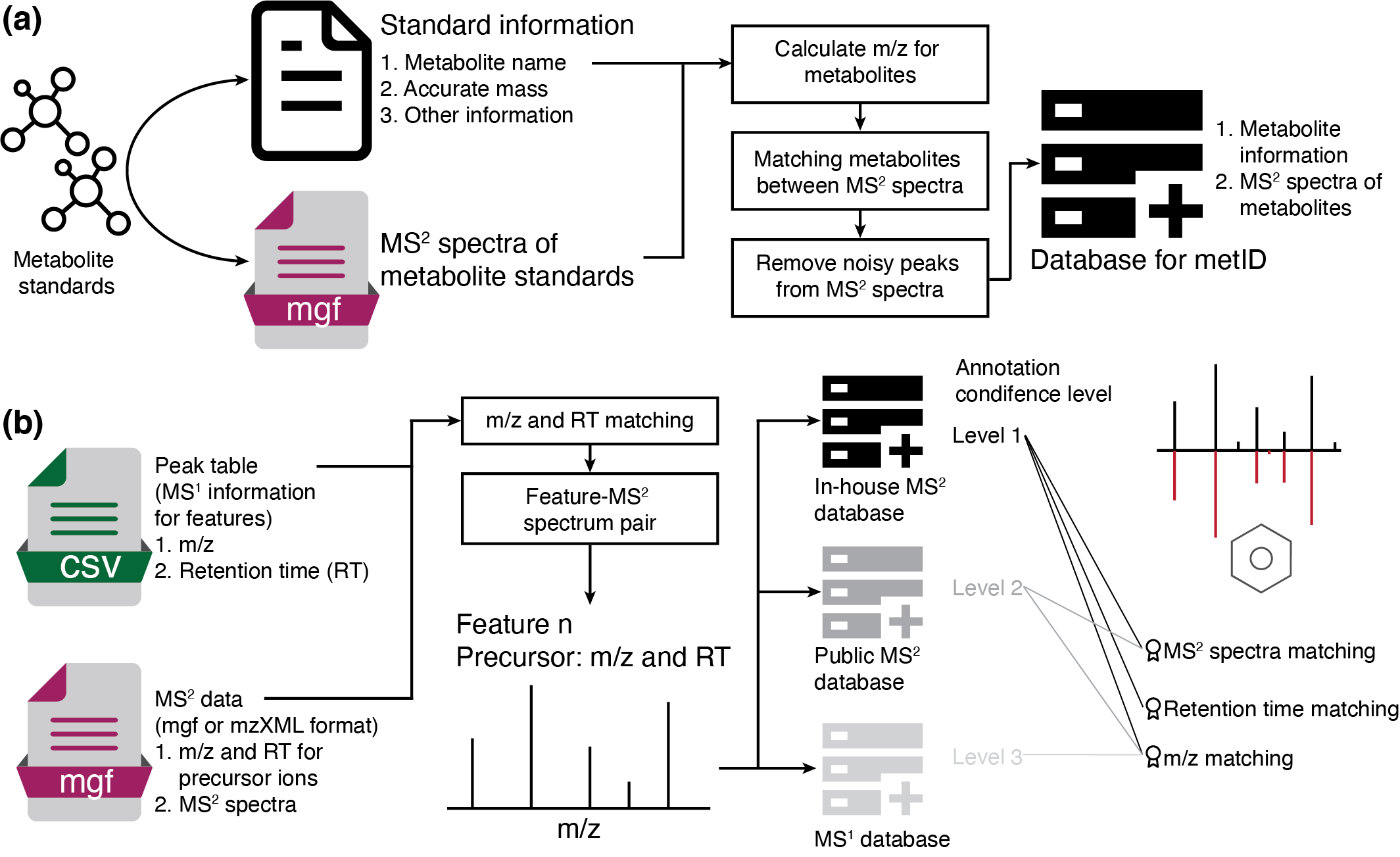
Installation
You can install metid from GitLab
if(!require(remotes)){
install.packages("remotes")
}
remotes::install_gitlab("tidymass/metid")or Github
remotes::install_github("tidymass/metid")metid is a part of tidymass, so you can also install it by installing tidymass.
Usage
Please see the Help documents page to get the instruction of metid.
Need help?
If you have any questions about metid, please don’t hesitate to email me (shenxt@stanford.edu).
{kind=link}
Citation
If you use metid in your publications, please cite this paper:
Xiaotao Shen, Si Wu, Liang Liang, Songjie Chen, Kevin Contrepois, Zheng-Jiang Zhu*, Michael Snyder* (Corresponding Author). metID: A R package for automatable compound annotation for LC−MS-based data. Bioinformatics, btab583, https://doi.org/10.1093/bioinformatics/btab583
Shen, X., Yan, H., Wang, C. et al. TidyMass an object-oriented reproducible analysis framework for LC–MS data. Nat Commun 13, 4365 (2022). Weblink
Thanks very much!